






Getting started with EADock
| Intro | Inputs | Outputs | Viewing Results | FAQ |
Introduction to EADock's concepts
EADock is a new protein/small ligand docking evolutionary approach implemented in Java, using CHARMM to handle structures and energy calculations.
From a scientific point of view, a sophisticated sampling algorithm, inspired by biological evolution, is combined with the well-know CHARMM package to score and manipulate conformations. This mixture not only works, but significantly outperforms other approaches.
From a technical point of view, EADock sends commands to an instance of CHARMM through its standard input, parsing on the fly its standard output, and monitoring its standard error for warnings and crashes (if any). With such a design, the power and flexibility of the object oriented design allowed in Java is combined with the speed of Fortran code.
Before going further with EADock, there is a couple of things to understand, and this page is here to help you getting started using EADock. Don't expect it to be accurate, as EADock is still under heavy development, but feel free to send me an email if you have any questions, suggestions or even complaints.
Overview of the algorithm
In a nutshell, EADock grows a population of docking poses. Usually, the initial docking poses are randomly generated. Over generation, the quality of the population increase, because docking poses are competing against each other with a selection pressure corresponding to energetic terms (SimpleFitness). The counterpart of this selection pressure involves several operators able to create new docking poses from the better poses identified in the population. During these generations, EADock carefully monitors the birth and death of docking pose clusters, reranking them according to a realistic scoring scheme (FullFitness, using GB-MV2). After several generations the evolutionary process stops.
Some of these steps are discribed briefly in the following sections.
Seeding
The seeding procedure is the generation of the initial population. EADock first reads usual PSF and a CRD files describing the complete system (Receptor+Ligand). This inital docking pose can be used to generate more or less controlled decoys to be incorporated in the intial population, or it can either be populated with a set of already generated docking poses.For more details, see below.
Then the evolutionary process starts.
Selection pressure
All docking poses (also called Agents) in the population are assigned a fitness regarding a selection pressure (SimpleFitness) based on their energy, and the fittest ones are automatically incorporated in the next generation.
Generation of diversity
To fill the remaining empty slots in the next generation, some of the fittest docking poses are modified by so-called Operators, with the hope that these modifications will lead to even better docking poses.
Post-operators
A local search can be performed, usually using minimisation, after the generation of a new docking pose, to speed up the search.
Diversity management
To avoid premature convergence, all generated docking poses are compared to existing ones in the population. This comparison is based on a RMSD clustering ensuring that a single docking mode will not infest the whole population. Several parameters are discussed below.
Printer
When the new population is full, it replaces the current one. At this point, special objects called Printer are used. They are responsible for generating a usable output from a population, such as a list of cluster, sets of coordinates, etc. Several Printers are available, see below.
Then, if the maximum number of generation has not been reached, a new evolutionary cycle restarts by a new selection process.
System partitioning
EADock has the ability to dock a ligand on the surface of a protein which can be either rigid, partly flexible, or completely flexible. To achieve this goal, the system is splitted in three subsystem defined by CHARMM selections:
Ligand |
corresponds to the ligand (must be SEGID LIG in the current implementation). This selection is explicitely modified by usual operators, and is kept flexible during post-operator minimisations. |
ReceptorFlex |
corresponds to the flexible part of the receptor. This selection can be modified by dedicated operators (not available yet), and is kept flexible during post-operator minimisations. |
ReceptorFix |
corresponds to the rigid part of the receptor, this is kept fixed to speedup energy calculations. |
These three selections are used by EADock to define the genome of a solution, fully describing a docking pose. This genome is composed of cartesian coordinates for the Ligand and ReceptorFlex selections.
Inputs
A sample docking run can be downloaded here. Refering to the files it contains while reading this document is likely to help you a lot in getting the most of EADock.
Files
EADock needs a parameter input file, together with several files. Some are optionals, others must be presents.
Required parameter files
| Suggested names | Content |
complex.crd |
CHARMM files describing an initial docking pose. |
sample.eadock |
EADock parameter file, with an awful syntax described in the following section. |
Optional parameter files
| Suggested names | Content |
par_all22_prot.inp |
CHARMM parameter and topology files, if you need to override the default ones bundled in EADock (taken from c31b1). |
extra.rtf |
Additional parameter and topology files for your system, if needed. |
lig.ic |
List of internal coordinates of the
ligand. This file is needed if CHARMM
is not able to reconstruct the ligand after part of it has been rotated
around bonds defined by the RotateBond
directive. This is usually the case for non peptidic ligands. This file
can be generated from complex.crd and complex.psf thanks to a
script that can be extracted from eadock.jar by the following command
(check the script header for its syntax):
unzip -p eadock.jar files/crdpsf2ic.prl >
crdpsf2ic.prl |
lig.sym |
Define symmetries of the ligand, listing equivalent combinations of heavy atoms. Taking symmetry into account leads to a more realisting clustering algorithm. Atoms are identified by their line numbering in the genome. This might require running EADock a first time to have a genome dumped to the disk, then list the equivalent atoms. Here, looking at the sample run is the best advice I can give. This file is not mandatory. If it's lacking or not specified in the directives of the parameter file, symmetries are ignored while calculating RMSD. |
ForceField.inp |
This file defines the force field parameters used
during the local search, using the CHARMM
syntax. You can specify what you need if
default
parameters are not suitable for your docking test. The default
is:NBOND CUTNB 14.0 CTOFNB 12.0 CTONNB 10.0 RDIE EPS 4.0 |
PostOperators.inp |
This file defined the local search method which is
applied after each Operator, using the CHARMM
syntax. You can specify what you
need if default
parameters are not suitable for your docking test. The default
is:MINI SD NSTEP 50 |
Parameter files syntax
Several parameters regarding both the docking procedure and the evolutionary algorithms have to be specified in so called "parameter files". Parameters are defined by single-line statements using a reserved word (the key, also called directive), followed by at least one space, and one or several coma-separated values. Double spacings are removed, and should have no impact. These directives are case sensitive, and lines starting with # are ignored. If the same directive is defined several time, only the last parsed value is kept. You need to specify a directive only if you do not want to override its default value. A list of available directives and their default values can be dumped by typing:
java -jar eadock.jar help
You can aslo generate a template parameter file that you can tweak according to your needs, with:
java -jar eadock.jar template > mytemplate.eadock
Altough it is
possible to gather the definition of all parameters in a single file,
splitting it might be convenient. For this purpose, the
Include directive can be used to load a
external parameter file (basically like "source" in bash) from the
current
parameter file. For example, if the main parameter file contains:Include
seeding.eadock
EADock will look for the file seeding.eadock and
read it as a parameter file before going further in the main parameter
file.
Paths to files
For each of this directive, more information is available in the file description section.
| Directive | Description |
PathPsfPathCrdReferenceStructure |
Define paths to the PSF/CRD of the complex used to start EADock (and sometimes to generate decoys to be used as seeds), and to the structure to use as a reference (to calculate RMSD to, usually an X-ray structure). |
PathCharmmTopologyPathCharmmParameter |
Define paths to CHARMM22 topology and parameter files. |
ForceFieldPostOperators |
Set paths to CHARMM input files defining the force field parameters, and the procedure applied after each Operator. |
GetLigandICFrom |
Defines the path to a file containing a list of internal coordinates for the ligand, to be sure that CHARMM will be able to reconstruct the whole ligand from any atom triplets defined as RotatableDihedral. |
ExtraRTF |
Defines the path to extra RTF and PAR files for the system. |
SymmetryFilename |
Define the path to a file containing several list of atom combinations to take symmetries into account in the ligand and/or in the flexible part of the receptor. This is used only when a reference structure has been defined. If this directive is not set, symmetries are simply ignored. |
Evolutionary algorithm parameters
| Directive | Description |
MaxGenerations |
Number of generations |
PopSize |
Number of docking poses competing in the population |
Replacement |
Number of Agents to replace at each generation. Other Agents are automatically copied to the next generations. |
AutoSchedulingMemoryDepth |
Automatic operator scheduling, the first directive should not be changed. The MemoryDepth sets the number of generations to take into account to recalculate operators probability. Thus, this also corresponds to the delay between two updates of these probabilities. |
MaxExpectedFitness |
If an Agent reach a fitness (-Energy) highest than this threshold, the evolutionary process stops. |
DistributionSubSystem |
Distributed computing facilities, not documented yet as it's my own private secret ;). |
GrowthType |
Should not be changed |
FitnessRescaling |
Should not be changed |
SaveEvery |
Should not be changed |
PrintEvery |
Should not be changed |
Seeding
Two alternative seeding mechanisms are available: automatic and manual. If none of the following directives are specified, the default seeding process is set to Automatic, without RMSD threshold (see below).
Automatic seeding
The seeding itself is performed by randomly rotating/translating the ligand position defined in the CRD file. Then, each RotateBond is optimized just like in the OptRot operator. Two thresholds can be specified to define a RMSD range of acceptance between the inital complex defined in the CRD and the generated conformation to be used as seed. Otherwise, there is no limits, meaning that the RMSD is not used to filter out docking poses populating the initial generation. These thresholds are defined with the following directives:
| Directive | Description |
MinRMSDToSee |
Minimal RMSD between the seed and the reference structure |
MaxRMSDToSeed |
Maximal RMSD between the seed and the reference structure |
Manual seeding
As the seeding is a CPU-intenstive task when dealing when several RotateBond or a big population, you can specify a zip file containing a set of CRD files containing coordinates for Ligand and ReceptorFlex, with the following directive:
Seeding ./structures.0-3.zip
Speeding up the seeding process.
EADock optimizes rotations and translations to try to generate conformations in the desired RMSD range. However, this can be highly inefficient for some Ligands, especially when they are not globular and/or have a lot of RotateBond.
An alternative is to generate decoys corresponding to the Ligand+ReceptorFlex parts of the system, then gather the corresponding CRD files into a single zip files. This zip file can then be declared to EADock using the Seeding directive:
Seeding ./my_own_seed_collection.zip
Operators
The Operator directive adds an operator to the evolutionary process.
Operator
<operator_class1>,<operator_class2>,...,<operator_classn>
Input/output parameters
Several directives can be used to generate a usable output, debug, and limit the needed disk space. There are two categories: general options, and Printers. See the output section for more information about the content of each file.
General options
| Directive | Description |
RemoteLog |
Redirect stderr to the EADock Error Collector running on a remote host. To be sure that the EADock Error Collector is running, you can send me a mail. |
DumpCharmmOutput |
Dump instruction sent to CHARMM, and CHARMM output. |
DumpBlacklistedDumpWhitelistedDumpRejected |
Dump blacklisted, whitelisted, and conformations rejected because of these black/whitelisted conformations in a zip file. |
Printers
The Printer directive adds a printer to the evolutionary process:
Printer <printer_class1>,<printer_class2>,...,<printer_classn>
Please note that the
sib.md.eadock.builders.ClusterManagerFakePrinterBuilder must be called
before other EADock specific printers.
Diversity management
EADock uses several tricks to manage a reasonnable diversity during the evolutionary process. This include: population on-the-fly clustering, limiting cluster size, cluster competition and reranking, choosing Operators according to the cluster to which the parent belongs to, and many others that I won't detail here.
Although most parameters are hardcoded, several directives are available to play with some of the degrees of freedom of these diversity management procedures:
| Directive | Description |
UseNicheRestrictionNicheDistanceCutoffNicheMaxSize |
Define whether EADock should limit cluster (or niche) size to prevent premature convergence. The RMSD distance cutoff defining a niche, and the maximum niche size can be specified here. |
ROI |
To limit the accessible search space, a region of interest can be defined. This ROI is defined with a distance criteria around the initial positioning of the Ligand. Two metrics are available: with this directive, distance corresponds to the distance between mass centers. Without this directive, the distance is the RMSD to the reference structure. |
MaxDistanceToROI |
Maximum distance to the ROI, either in RMSD, or in Angströms. |
UseOperatorRangeForNicheSampling |
Should not be changed |
EvaluateClusterRankMethod |
Should not be changed |
UniqueOnly |
Should not be changed |
LimitClusterSizeBlackListingStartBlacklistingWhiteListingStartWhitelistingFinalReranking
trueFinalRerankingMinimizationStepsFinalRerankingClusterNumberAcceptanceThresholdAllowStopAtStopIfNicheRadiusBelow |
Turns on/off niche restriction, blacklisting, whitelisting, and the final reranking of n clusters after a given number of GB-MV2 miminization steps. It also defined when the algorithm will be able to stop when blacklisting/whitelisting lead to a too small yield of creating acceptable new solutions, as well as the yield itself. This should not be changed. |
Fitness parameters
| Directive | Description |
StartIntraFullIntraStartFullFullFull |
Should not be changed |
FitnessSelection |
Using RMSD or RANDOM instead of REAL will replace the REAL (energy-based) driving force by either a RMSD-based or a RANDOM driving force. This is useful for assessing the algorithm assessment, but useless otherwise. |
DedicatedCHARMMForGBMV |
Use a dedicated CHARMM instance for single point GB-MV2 calculations. This is likely to help when c31b1 is used, as its implementation of GB-MV2 has some bugs. |
Testcase dependant section
| Directive | Description |
EadockSessionName |
Sets the name for the current session. This name is used to create the output directory, as well as some output files. |
LigandReceptorFlexReceptorFix |
CHARMM selections for the Ligand, the receptor flexible part, and the receptor fixed part. Note that the "SELE" and the "END" keywords must not be used in these lines. |
Rotatable bonds definition
The RotateBond is used to define bonds that are explicitely (and systematically) rotated by the OptRot Operator. A coma must be present to separate several bond definitions, which consist of two atoms. Each atom is identified by two elements: the numbering of the residue of the ligand to which the atoms belongs to, and the atom name.
RotateBond 1 C1' 1 N9, 1 C4' 1 C5'
Outputs
EADock stores all its output in a directory called <EadockSessionName>.<version>. If a directory exists with the same name, the counter <version> is appened, incremented until the desired name is available. During a run, depending on the Printers you selected, EADock will create several output files and directories.Output files
| File | Content | Directive |
<EadockSessionName>.<version>/ea.param |
copy of the parameters for the current run | - |
<EadockSessionName>.<version>/<EadockSessionName>.out |
Almost useless, prints out the fitness of all Agent for all generations, together with the memory used, the evolution of operators probabilities (with automatic operator scheduling, etc.) | Redirect |
<EadockSessionName>.<version>/<EadockSessionName>.err |
This file shows what's happening during the run. It
contains CHARMM
warnings as well as EADock specific informations about the current step
of the algorithm (creating or evaluating an docking pose, clustering
the population and managing
its diversity, etc.). Each line starts with the object sending the message, a semi-colon, and the message itself. |
Redirect |
Conformations impacted by the diversity management system
Three files contain conformations selected or rejected by the diversity management.| File | Content | Directive |
<EadockSessionName>.<version>/blacklisted.zip |
contains conformations discarded because of their full score. | DumpBlacklisted |
<EadockSessionName>.<version>/whitelisted.zip |
contains conformations around which the search must be focused. | DumpWhitelisted |
<EadockSessionName>.<version>/rejected.zip |
contains conformations rejected because they are either too close from a blacklisted conformation, or too far from a whitelisted conformation. | DumpRejectedlisted |
Debug files
| File | Content | Directive |
<EadockSessionName>.<version>/<EadockSessionName>.charmm.in |
commands streamed to CHARMM | DumpCharmmOutput |
<EadockSessionName>.<version>/<EadockSessionName>.charmm.out |
CHARMM output. | DumpCharmmOutput |
Conformations
The "pdb" directory
All conformations generated and evaluated are stored in the "pdb" directory. For each generation, two files are created:| File | Content | Directive |
<EadockSessionName>.<version>/pdb/g<Generation>.crd.zip |
contains all conformations (CRD format). | Printer
sib.md.eadock.builders.PDBPrinterBuilder |
<EadockSessionName>.<version>/pdb/g<Generation>.clusters.csv |
list identified clusters features. | Printer
sib.md.eadock.builders.ClusterPrinterBuilder |
The "results" directory
This is where your most favorable docking modes are stored.
After the last generation, the population is clustered. For each best 10 clusters, the conformation with the lowest energy is minimized using GB-MV2 (250 steps ABNR) and the coordinated after minimization are written in this directory. This require the FinalReranking directive to be set to true (default=false).Viewing results
You can both look at minimized conformation from the result directory, or load whole generations. The visualization program called Chimera (download) have the nice ViewDock tool. Here is a script to converting the zip file containing all agents of a generation into a Dock4 result file: zip2dock4 (this script requires another script, crd2pdb.prl, to be in the path).Load inital conformation
First load the initial conformation in Chimera.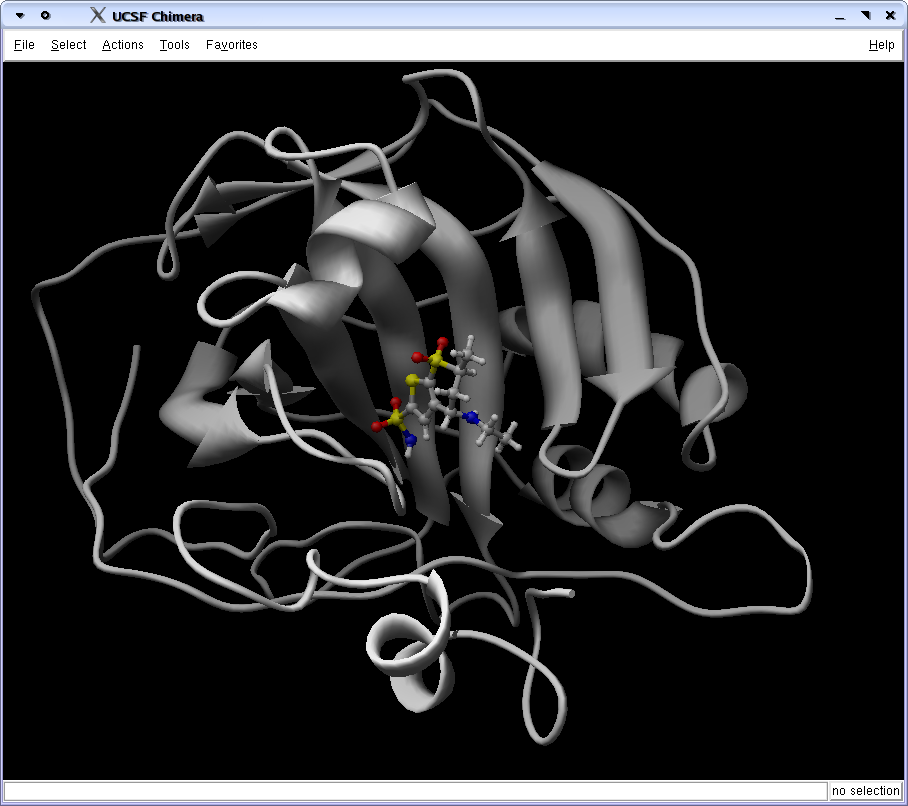
Load docking results
Then go in Tools>Docking>ViewDock, and select the dock4 file you created a few line before, and load it as a DOCK4 file. You can then add EADock specific tags such as RMSD to reference or fitness by selecting them in the Column>Show menu. On the following picture, the cluster with the lowest energy is pictured in magenta, together with the X-ray conformation colored according to atom types.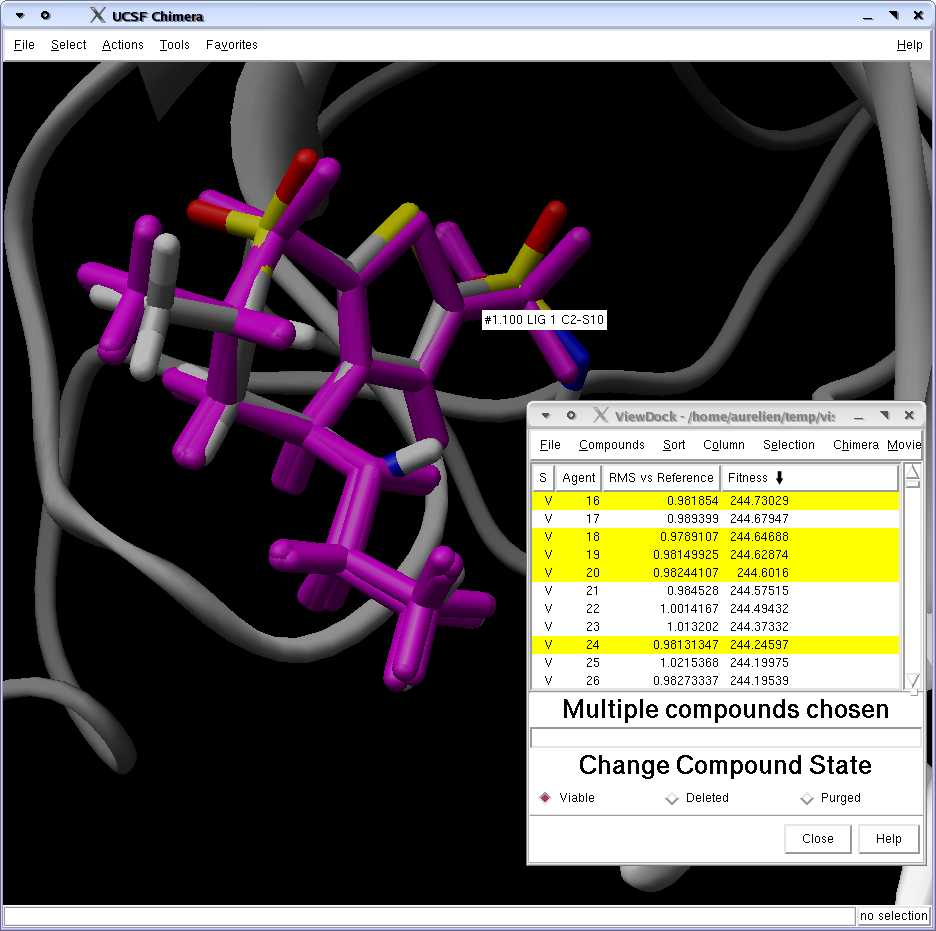
FAQ
Crash and solutions
Both CHARMM and EADock might crash. You must first identify which one had a problem. Looking at the end of the error file will help a lot. You can either see a list of exceptions thrown by the java virutal machine (this is an EADock crash), or a line "the CHARMM instance has exited" followed with the last 1000 lines of the CHARMM output.| Crash | Solution |
| EADock | First read the Java error, and try to understand what could be the problem. Very often, such crashes are related to wrong input parameters. Then check your parameters, then you might eventually send me mail so that i can investigate a bit. |
| CHARMM | Look at the 1000 last lines of CHARMM's output to understand why it crashed. Note that the GB-MV2 code is maybe not so bulletproof, and i'm really not the guy to report GB-MV22 bugs to. |
java -Xmx512m -jar eadock.jar <eadock input
file>Note: you must use c28a4 or newer to avoid deadlock, and c31a1 to use GB-MV22 !
Speed problem
If the evolutionary process seems to slow down, or even to stop, a simple top is very likely to help you. CHARMM should take most of the CPU time. If java is eating everything, there is probably an infinite loop in EADock, and you should consider killing the current run. Checking the time used by the CHARMM process, compared to the time used by the Java process will help you a lot. Note: as there are some peaks of java activity, for instance during the clustering, looking at the CPU usage is probably not that relevant.
If CHARMM seems to be calculating something while the evolutionary process is very slow, looking at the last lines of the error file is tell you what it is currently calculating. Note that EADock is very time-consuming, especially the automatic seeding and the last minimizations using GB-MV2. A typical docking run with a ligand containing 10 RotateBond and a receptor of 300aa is likely to take >12 hours. Look at the timestamps of files created at each generation to check if the slow down you are observing is relevant or not.


